Euroopan geneeristen lääkkeiden sääntely: Käytännön opas EU:n reitteihin ja uudistuksiin
 kesä, 10 2026
kesä, 10 2026
Geneeristen lääkkeiden saatavuus Euroopassa ei ole yhtä suoraviivaista kuin moni kuvittelee. Vaikka tavoitteena on edullisempi hoito kaikille 27 jäsenvaltion asukkaille, käytännössä valmistajat joutuvat navigoimaan labyrinttimaisen säätelyjärjestelmän läpi. Vuoden 2025 lääkepaketti (EU Pharma Package) toi mukanaan muutoksia, jotka muuttavat pelin sääntöjä - erityisesti tietosuojakausien lyhentymisen ja toimittamisvelvoitteiden tiukentumisen myötä.
Tämä artikkeli purkaa solmua siitä, miten geneeristen lääkkeiden hyväksyntä todella tapahtuu EU:ssa. Olemme katselleet eri hakumenettelyjen eroja, kustannuksia ja aikatauluja sekä tarkastelleet, mitä vuoden 2025 ja 2026 uudistukset tarkoittavat konkreettisesti yrityksille ja potilaille. Tavoitteena on antaa selkeä kuva siitä, miksi jonkin lääkkeen saatavuus voi viivästyä kuukausilla tai jopa vuosilla riippuen valitusta reitistä.
Miksi EU:n geneerilääkejärjestelmä on niin monimutkainen?
Euroopan unionin lääkesääntely perustuu tasapainotteluun turvallisuuden ja kilpailun välillä. Geneeriset lääkkeet ovat lääkkeitä, jotka sisältävät saman vaikuttavan aineen kuin alkuperäinen lääke, mutta joita myydään usein alhaisemmalla hinnalla. Ne muodostavat noin 65 % kaikista EU:ssa reseptoiduista lääkkeistä tilavuudeltaan, vaikka niiden arvo on vain 18 % kokonaismarkkinoista (EGA, 2024). Tämä rako korostaa sitä, kuinka kriittinen rooli geneereillä on terveydenhuollon kustannusten hallinnassa.
Järjestelmän ydinongelma on ollut sen hajanaisuus. Vaikka Euroopan lääkevirasto (EMA European Medicines Agency) harmonisoi sääntöjä, lopullinen päätösvalta markkinoinnin sallimisesta on usein kansallisilla viranomaisilla. Tämä on aiheuttanut tilanteen, jossa sama lääke voi olla saatavilla Saksassa, mutta puuttua Hollannista tai Belgiasta kuukausien ajan teknisten hyväksyntöjen jälkeen. Euroopan komission vaikutusarvioinnin (2023) mukaan tämä fragmentaatio viivästyttää geneerien saantia keskimäärin 11,3 kuukautta jäsenvaltioiden välillä.
Neljää tietä markkinoille: Kumpi sopii parhaiten?
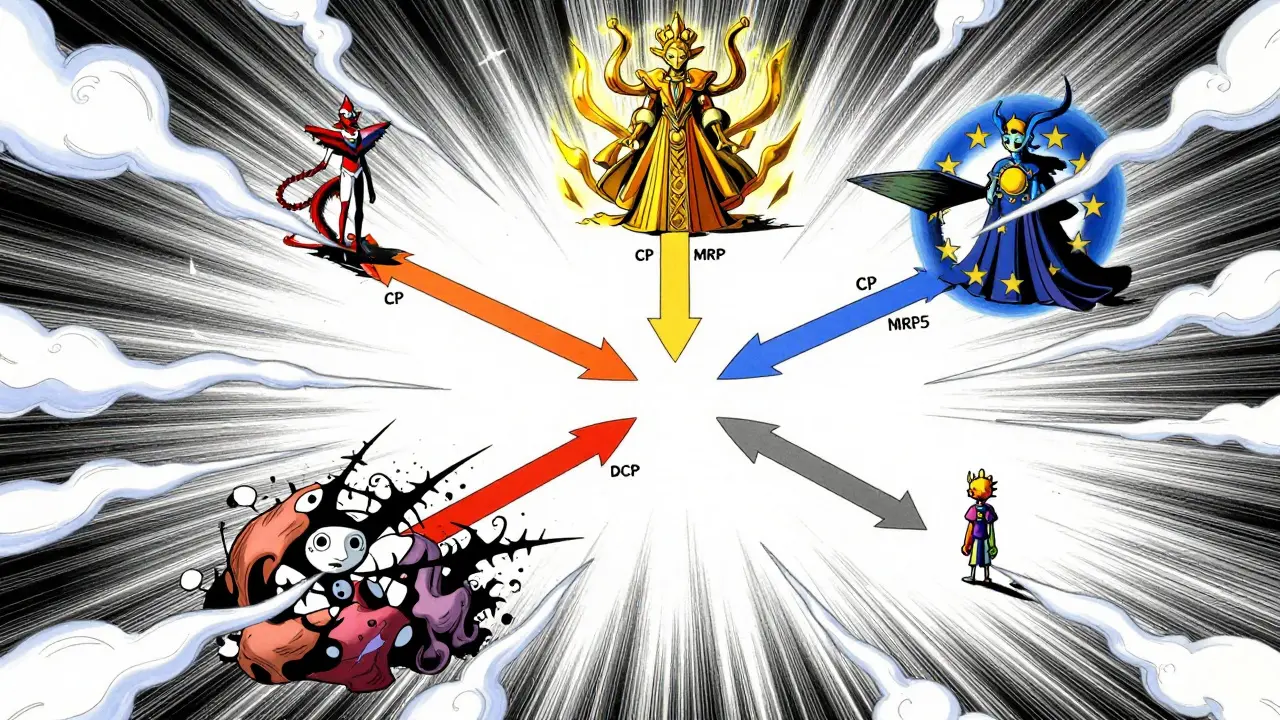
Kun yritys haluaa tuoda geneerilääkkeen markkinoille, se joutuu valitsemaan yhdestä neljästä hyväksyntäreitistä. Valinta riippuu lääkkeen arvosta, kohdemarkkinoista ja yrityksen resursseista. Jokaisella reitillä on omat nopeutensa, kustannuksensa ja riskinsä.
- Keskitetty menettely (Centralized Procedure, CP): Tämä on suoraviivaisin reitti laajalle levikille. Hakemus jätetään suoraan EMA:lle, ja myöntämispäätös pätee kaikissa 27 EU-maassa sekä Islannissa, Liechtensteinissa ja Norjassa. Tieteellinen arviointi kestää nykyisin 210 päivää, mutta vuoden 2025 uudistuksen nojalla se lyhenee 180 päivään. Prosessi on kallis - sovellusmaksut nousevat noin 425 000 euroon ja konsulttikustannukset 1,2-1,8 miljoonaan euroon (PwC, 2024). Siksi se kannattaa pääasiassa korkean arvon lääkkeille, joiden odotetaan tuottavan yli 250 miljoonaa euroa vuodessa.
- Väestövälisen tunnustamisen menettely (Mutual Recognition Procedure, MRP): Noin 42 % geneerihakemuksista kulkee tämän reitin. Lääke hyväksytään ensin yhdessä maassa (viitejäsenvaltiossa), ja sitten muut maat tunnustavat päätöksen. Teoriassa yhteisymmärrys pitäisi syntyä 90 päivässä, mutta käytännössä se vie keskimäärin 132,7 päivää kansallisten erimielisyyksien vuoksi (IQVIA, 2024). Alkukustannukset ovat matalammat (180 000-220 000 euroa), mutta riskinä on, että hintoneuvottelut yhdessä maassa (kuten Saksassa) voivat hidastaa myyntiä muualla.
- Dekentralisoitu menettely (Decentralized Procedure, DCP): Tällä reitillä, jota käyttää 38 % hakijoista, haetaan lupaa useaan maahan samanaikaisesti ilman aiempaa kansallista lupaa. Viitejäsenvaltio johtaa arviointia. Vaikka aikaraja on 210 päivää, prosessi venyy keskimäärin 247 päivään koordinaatiovaikeuksien vuoksi. Erityisesti Itä-Euroopan maiden tulkinnat laadunvarmistusvaatimuksista vaihtelevat, mikä aiheuttaa merkittäviä viiveitä.
- Kansallinen menettely (National Procedure): Vain 5 % hakemuksista käyttää tätä reittiä. Se on tarkoitettu yksittäisiin maihin suunnatuille tuotteille. Prosessi kestää 180-240 päivää eikä hyödynnä EU:n harmonointia. Esimerkiksi Accord Healthcare:n kokemus Ranskassa osoitti, että kansallinen lupa vei 197 päivää, kun taas MRP-reitti viidelle muulle maalle olisi voinut olla tehokkaampi.
| Menettely | Kesto (keskim.) | Arvioitu kustannus | Soveltuvuus |
|---|---|---|---|
| Keskitetty (CP) | 180-210 pv | €1,6-2,2 milj. | Korkean arvon lääkkeet, koko EU |
| Väestöväläinen (MRP) | ~133 pv | €180-220 t. | Päämarkkina + muut maat |
| Dekentralisoitu (DCP) | ~247 pv | Muuttuva | Useat maat, ei etukäteislupaa |
| Kansallinen | 180-240 pv | Matala | Yksi maa |
Sääntelyllinen ekvivalenssi: Mitä todistaa?
Riippumatta valitusta reitistä, geneerilääkkeen on oltava terapeuttisesti ekvivalentti alkuperäislääkkeen kanssa. Tämä ei ole vain väite vaan tiukka tiedollinen vaatimus. Valmistajan on osoitettava, että lääkkeessä on sama laadullinen ja määrällinen koostumus vaikuttavista aineista, sama farmaseuttinen muoto ja että se täyttää bioekvivalenssivaatimukset.
EMA:n ohjeistuksen mukaan bioekvivalenssitutkimusten tulosten 90 % luottamusvälin on sijoittua alueelle 80,00-125,00 % sekä Cmax- (huippukonsentraatio) että AUC- (kokonaisaltistus) parametreille. Monimutkaisemmissa geneereissä, kuten inhalaattoreissa, vaatiot kiristyvät. Esimerkiksi Saksan BfArM vaatii usein lisätestejä farmakodynaamisista vaikutuksista, vaikka EMA:n yleiset standardit olisivatkin täyttyneet. Tämä on yksi syy siihen, miksi 68 % geneerivalmistajista pitää kansallisia bioekvivalenssitulkintoja suurimpana esteenä (ABPI, 2025).
Vuoden 2025 ja 2026 uudistukset: Mitä ne tarkoittavat sinulle?
Lääketeollisuudessa puhutaan paljon vuoden 2025 lääkepaketista (Pharma Package), joka astui voimaan vaiheittain. Sen vaikutukset näkyvät nyt ja tulevaisuudessa kolmella keskeisellä tavalla:
- Bolar-poikkeuman laajentaminen: Aiemmin geneerivalmistajat saattoivat aloittaa hintaneuvottelut vain kaksi kuukautta ennen patentin vanhenemista. Nyt ikkunaa on laajennettu kuuteen kuukauteen. REMAP Consultingin mallinnuksen (2025) mukaan tämä voi nopeuttaa markkinoilletuloa keskimäärin 4,3 kuukautta ja laskea hintoja 12-18 % aiemman kilpailupaineen ansiosta.
- Tietosuojakauden lyhentäminen: Perinteinen 10 vuoden tietosuoja (8+1) lyhenee. Uusi malli tarjoaa 8 vuotta tietosuojaa ja 1 vuoden markkinasuojaa, jota voidaan pidentää enintään 2 vuoteen, jos lääke täyttää tietyt julkisterveyshankkeet. Tämä muutos astuu voimaan heinäkuussa 2026 ja saattaa kiihdyttää geneerien saantia erityisesti biologioiden osalta.
- Toimittamisvelvoite (Obligation to Supply): Uudet säännöt pakottavat valmistajia varmistamaan riittävät lääkemäärät. Professori Panos Kanavos varoittaa kuitenkin, että kansallisten viranomaisten erilaiset tulkinnat "riittävästä määrästä" voivat luoda keinotekoisia pullokauloja pienemmillä markkinoilla.

Käytännön haasteet ja kustannukset
Teoreettiset aikataulut poikkeavat usein todellisuudesta. Mylan (nykyisin Viatris) raportoi vuonna 2024, että MRP-prosessin viiveet lisäsivät kantokustannuksia 3,2 miljoonalla eurolla per korkean arvon lanseeraus. Lisäksi uusien sähköisten tuotetietojen (ePI) XML-muotoisten ilmoitusten velvoite, joka tulee voimaan vuonna 2026, vaatii IT-infrastruktuuriinvestointeja, jotka arvioidaan olevan 180 000-250 000 euroa yritystä kohti (White & Case, 2025).
Huolimatta haasteista, strateginen valinta maksaa itsensä takaisin. Sandoz hyödynsi keskitettyä menettelyä Novartis Cosentyx -lääkkeen geneeriversiossa, saavuttaen koko EU:n kattavan lanseerauksen Q2 2025 - 11 kuukautta nopeammin kuin perinteinen MRP olisi mahdollistanut. Tämä esimerkki korostaa, että oikea reittivalinta on ratkaisevaa liiketoiminnan kannattavuudelle.
Markkinatilanne ja tulevaisuuden näkymät
EU:n geneerilääkemarkkinoiden arvo oli 42,7 miljardia euroa vuonna 2024, kasvaen 6,2 % edellisvuodesta. Keski- ja Itä-Eurooppa kasvavat nopeimmin, 9,8 % vuositasolla (IQVIA, 2025). Kilpailu kiristyy, kun Intian valmistajat ottivat osakseen 38 % EU:n geneeriluvista vuonna 2024, kun taas eurooppalaiset jättit kuten Sandoz ja Viatris pitävät 52 % markkinaosuudestaan keskittyen korkean arvon segmentteihin.
Critical Medicines Act (maaliskuu 2025) tuo lisää vakautta pakottamalla 200 elintärkeän geneerin varastoinnin, mutta se nostaa myös kynnystä markkinoille uudenlaisten laaduntarkastusprotokollien kautta. Yhdysvaltojen ja EU:n välinen kehyssovinno (syyskuu 2025) saattaa vaikuttaa raaka-aineiden tullikohteluun, mikä voi muuttaa valmistuskustannuksia lähitulevaisuudessa.
Mitä eroa on keskitetyllä ja dekesentralisoidulla menettelyllä?
Keskitetyssä menettelyssä hakemus jätetään suoraan EMA:lle, ja lupa pätee kaikissa EU-maissa samanaikaisesti. Dekentralisoidussa menettelyssä hakemus jätetään useaan maahan kerralla, mutta viitejäsenvaltio johtaa arviointia ja muut maat seuraavat sen jäljessä, mikä voi aiheuttaa viiveitä.
Milloin vuoden 2025 lääkepaketin muutokset astuvat täysin voimaan?
Muutokset tulevat voimaan vaiheittain. Bolar-poikkeuman laajentuminen astui voimaan syyskuussa 2025, ja keskeinen tietosuojakauden lyhentäminen (8+1 malli) astuu voimaan heinäkuussa 2026.
Miksi geneerilääkkeen saatavuus voi viivästyä EU:ssa?
Viiveet johtuvat usein kansallisten viranomaisten erilaisista tulkinnista laatuvaatimuksista, hintaneuvotteluista tai toimittamisvelvoitteiden täyttämisestä. Fragmentoitunut järjestelmä voi viivästyttää saantia keskimäärin yli 11 kuukaudella jäsenvaltioiden välillä.
Kuinka paljon geneerilääkkeen hyväksyntä maksaa?
Kustannukset vaihtelevat valitusta reitistä. Keskitetty menettely voi maksaa yli 1,6 miljoonaa euroa kaikkine kuluisineen, kun taas väestövälisen tunnustamisen (MRP) alkukustannukset ovat noin 180 000-220 000 euroa. Kansallinen menettely on halvin, mutta se rajoittaa myynnin yhteen maahan.
Mitä bioekvivalenssi tarkoittaa käytännössä?
Bioekvivalenssi tarkoittaa, että geneerilääke imeytyy elimistöön samalla tavalla ja määrin kuin alkuperäislääke. Tutkimusten tulosten on sijoittuttava 80-125 % luottamusväliin verrattuna referenssilääkkeeseen.
Sanna Hiltunen
kesäkuu 12, 2026 AT 05:41kiitos tästä erinomaisesta kokonaisuudesta joka todella avaa silmiä monelle meistä jotka työskentelevät terveydenhuollon alalla ja kohtaamme näitä byrokraattisia esteitä päivittäin
minusta on hienoa että viimein huomioidaan se fragmentaatio ongelma joka on hidastanut geneerilääkkeiden saantia niin paljon eri jäsenvaltioissa
olen itse kokenut miten sama lääke voi olla saatavilla Saksassa mutta puuttua täysin Suomesta vaikka kyseessä olisi elintärkeä hoitomuoto
vuoden 2025 uudistukset kuulostavat lupaavalta erityisesti tietosuojakauden lyhentäminen joka toivottavasti nopeuttaa biologioidenkin kilpailutilannetta
toivoisin että nämä muutokset toteutuisivat myös käytännössä niin tehokkaasti kuin paperilla näyttää olevan suunniteltu
on tärkeää että valmistajat saavat selkeät ohjeet joita he voivat noudattaa ilman turhia tulkinta-eroja kansallisten viranomaisten välillä
bioekvivalenssivaatimusten yhtenäistäminen olisi myös valtava askel oikeaan suuntaan koska nyt jokainen maa vaatii hieman eri asioita
kustannuslaskelmat ovat mielenkiintoisia ja ne korostavat sitä miksi pienemmät yritykset joutuvat usein valitsemaan tietyt reitit tarkemmin
keskitetty menettely on kallista mutta joskus ainoa järkevä vaihtoehto laajalle markkina-alueelle
uskon että tämä artikkeli auttaa monia ymmärtämään paremmin mitä taustalla tapahtuu kun lääkeviiveitä ilmenee
olisi hyvä jos saisimme enemmän dataa siitä miten Bolar-poikkeuman laajentaminen vaikuttaa konkreettisesti hintoihin potilaiden näkökulmasta
terveydenhuollon kustannusten hallinta on kriittistä ja geneeriset lääkkeet ovat siinä keskeisessä roolissa
kiitos vielä kerran tämän laadukkaan analyysin jakamisesta yhteisölle
Sune Törnqvist
kesäkuu 12, 2026 AT 08:10vau mikä labyrintti sääntöjen maailmassa
tuo keskitetty menettely maksaa ihan pahan rahat mutta ehkä se on silti sen arvoista jos halutaan välttää ne kansalliset kiemurat
mielenkiintoista että MRP-reitti on niin yleinen vaikka se venyy keskimäärin yli neljän kuukauden ajan
ehkä pitäisi luottaa siihen että EMA osaa hommansa paremmin kuin yksittäiset maat
mutta sitten taas ne hintaneuvottelut Saksassa hidastavat kaikkea muuta
ihmeellinen systeemi totta kai
kaisu marjatta
kesäkuu 12, 2026 AT 17:52byrokratia tappaa inhimillisyyden
lääkkeet ovat ihmisoikeus ei pelkkää liiketoimintaa
miksi meidän pitää odottaa vuosia että saadaan edullinen versio lääkkeestä
systeemi on rikki
Mattias Köhlmark
kesäkuu 12, 2026 AT 21:19EU:n sääntely on aina liian kireää ja se tuhoaa paikallisen teollisuuden
meidän pitäisi pystyä päättämään omista lääkkeistämme ilman Brysselin hässäkkää
ne halpa intialaiset tuotteet tulvivat markkinoille ja eurooppalaiset yritykset kärsivät
on aika ottaa takaisin kontrolli omasta terveydenhuollostamme
ei tarvitse kaikkia 27 maata samanaikaisesti vaan jokainen hoitaa oman asiansa
sekava kama tuo koko paketti
Britt Morrison
kesäkuu 13, 2026 AT 01:00hei kaikki
olen hyvin utelias kuulla lisää siitä miten nämä uudistukset vaikuttavat käytännössä potilaitten arkeen
onko jollakin kokemuksia viiveistä joita mainittiin artikkelissa
mitä tarkoittaa se että tietosuoja lyhenee biologioiden kohdalla
haluaisin ymmärtää paremmin miten tämä nopeuttaa saatavuutta
onko ollut esimerkkejä joissa lääke on tullut markkinoille aiemmin kuin ennen olisi ollut mahdollista
kuuntelen mielelläni ajatuksianne asiasta
Kristina Lund Hansen
kesäkuu 15, 2026 AT 00:50vaikka artikkeli näyttää neutraalilta se unohtaa mainita tärkeimmän asian eli voitonjako
geneerivalmistajat eivät tee tätä hyväntekeväisyyteen vaan rahaksi
uudet säännöt vain siirtävät kustannuksia eri toimijoiden väliin eivätkä ne välttämättä laske lopullista hintaa potilaalle
on naiivia uskoa että byrokratian vähentäminen automaattisesti tarkoittaa halvempia lääkkeitä
usein ne säännöt on tehty suurten farmaseuttisten yritysten eduksi
joten kannattaa olla varovainen optimististen ennusteiden kanssa
Malin Algotsson
kesäkuu 15, 2026 AT 14:27heippa Britt ja Kristina
on todella tärkeää että puhumme näistä asioista ääneen koska terveys on perusoikeus
malinin näkökulmasta on moraalisesti väärin että ihmiset joutuvat odottamaan hoitoa byrokratian vuoksi
meidän on velvollisuutena vaatia parempaa järjestelmää joka palvelee ihmisiä ei vain yrityksiä
toivon että vuoden 2025 uudistukset tuovat oikeasti muutosta ja että emme unohda niitä haavoittuvia ryhmiä
yhteisöllisyys ja huolenpito ovat avainasemassa tässä keskustelussa
pitääkö meidän odottaa että joku muu ratkaisee ongelmat vai voimmeko me vaikuttaa
uskon että yhdessä voimme tehdä eroa
Kristina Davies
kesäkuu 16, 2026 AT 18:32te ette ymmärrä markkinataloutta
jos hinta on korkea niin se on koska tuotanto on kallista
geneerit ovat halpoja koska ne hyödyntävät alkuperäisen tutkimustyötä
mutta jos poistatte suojat liian aikaisin niin innovaatiot lakkaavat
on tasapaino löydettävä eikä vain poliittisia iskulauseita huudeta
tiedän tarkalleen miten nämä prosessit toimivat ja ne ovat monimutkaisia
älä ole niin naivi että luulet että kaikki on mustavalkoista
Sven Hartge
kesäkuu 17, 2026 AT 11:40ei minua huvita lukea näitä pitkiä tekstejä
olen uupunut kaikesta sääntelystä ja byrokraatiasta joka vie aikaa todelliselta työltä
teidän pitäisi keskittyä siihen että lääkkeet toimii eikä siihen miten ne hyväksytään
olen väsynyt kuulemaan samoja valituksia joka päivä
jätä minut rauhaan ja anna minun tehdä oma työni ilman näitä häiritseviä keskusteluja
olen pettynyt tähän foorumiin koska se ei tuo mitään uutta
Benny Jensen
kesäkuu 18, 2026 AT 01:33ymmärrän Svenin tunteet ja kuuntelen sinua
on raskasta kantaa vastuuta kun systeemi tuntui jumittuvan
haluan vain sanoa että olen täällä sinun kanssasi ja kuulen sinut
toivottavasti löydät hetken rauhaa ja lepoa keskellä kaikkea this chaos
sinun hyvinvointisi on tärkeä ja pidän sinusta
Björn M I
kesäkuu 18, 2026 AT 18:48suomi ja ruotsi ovat aina olleet erillisiä entiteettejä ja meidän ei pitäisi alistua EU:n painostukseen
meidän pitäisi pitää omat lääketuotteemme ja omat sääntömme
tuota dekesentralisoitua menettelyä käytetään liian harvoin vaikka se olisi meille parempi
haluan että Suomi päättää omista asioistaan ilman ulkopuolisia vaikutteita
on häpeällistä että luotamme ulkomaisiin valmistajiin niin paljon
pitäisi tukea kotimaista teollisuutta enemmän